FDA niet transparant over sterfgevallen geassocieerd met via catheter ingebrachte hartkleppen
 Op 24 december 2019 verscheen op Kaiser Health News een interessant artikel met behoorlijke implicaties. Christina Jewett schreef “Reports Of Patients’ Deaths Linked To Heart Devices Lurk Below Radar”. In het artikel maakt zij duidelijk dat de controlerende Food and Drug Administration(FDA), gebruik maakt van een rapporteringssysteem dat haar onvoldoende zicht geeft op die data. Het gaat om via de bloedbaan m.b.v. een katheter ingebrachte hulpmiddelen in het hart of grote vaten. Het betreft op deze wijze verrichtte hartklep-vervanging, hartklep-aanpassing en vaatverwijding. De overlijdensgevallen die daaraan gerelateerd zijn zitten in registratiesystemen die vallen onder een “registry exemption program” van de FDA. De vorm van rapporteren is zodanig dat het zowel voor de FDA als voor onderzoekers en publiek het tot twee jaar kan duren om inzage te krijgen. Overlijdensrapporten t.g.v. “medical devices” horen open te zijn voor onderzoekers om hun collega’s te kunnen volgen en te waarschuwen voor veiligheidsproblemen.
Op 24 december 2019 verscheen op Kaiser Health News een interessant artikel met behoorlijke implicaties. Christina Jewett schreef “Reports Of Patients’ Deaths Linked To Heart Devices Lurk Below Radar”. In het artikel maakt zij duidelijk dat de controlerende Food and Drug Administration(FDA), gebruik maakt van een rapporteringssysteem dat haar onvoldoende zicht geeft op die data. Het gaat om via de bloedbaan m.b.v. een katheter ingebrachte hulpmiddelen in het hart of grote vaten. Het betreft op deze wijze verrichtte hartklep-vervanging, hartklep-aanpassing en vaatverwijding. De overlijdensgevallen die daaraan gerelateerd zijn zitten in registratiesystemen die vallen onder een “registry exemption program” van de FDA. De vorm van rapporteren is zodanig dat het zowel voor de FDA als voor onderzoekers en publiek het tot twee jaar kan duren om inzage te krijgen. Overlijdensrapporten t.g.v. “medical devices” horen open te zijn voor onderzoekers om hun collega’s te kunnen volgen en te waarschuwen voor veiligheidsproblemen.
Delegeren
Dat “registry exemption program” van de FDA is een vorm van delegatie van verantwoordelijkheden richting de markt. Ik schreef over speciale vormen van delegatie van verantwoordelijkheden door de FDA al eerder over op 30 december 2019. De nu besproken regeling is in 2010 in gang gezet. Overlijdensgevallen die verband houden met de per catheter ingebrachte “medical”devices hoefden dan niet rechtstreeks aan de FDA gerapporteerd te worden, maar richting particulier medische verenigingen. Die leveren de data in spreadsheets aan bij de fabrikanten. Vervolgens leveren die de data aan bij de FDA. Volgens standaard FDA-rapportageregels is de fabrikant verplicht die sterfgevallen te onderzoeken. Men moet dan een gedetailleerd openbaar rapport sturen over elk apparaat- gerelateerd overlijdensgeval. De fabrikanten die onder het “exemption” programma werken stellen dat de aanleverende organisaties in de verslaglegging sleutelgegevens(persoonskenmerken) verwijderen. Gegevens die de fabrikanten nodig hebben om elke dood volledig te onderzoeken.
Hoofdmoot
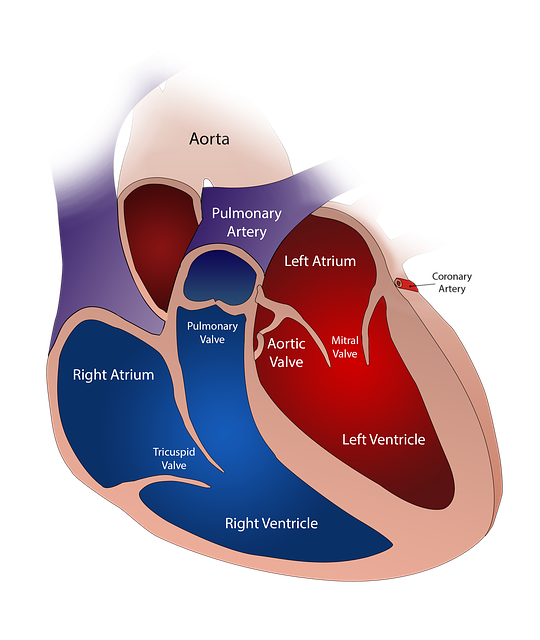
Hoofdzakelijk gaat het bij de onder deze regeling vallende fabrikanten om hartkleppen die m.b.v. een katheter ingebracht worden om een defecte te vervangen. In haar artikel gaat Christina Jewett uitgebreid in over de registratiepraktijk van mensen waarbij het overlijden gerelateerd was aan het inbrengen van een hartklep via de vaten. Ze noemt daarbij expliciet de CoreValve van Medtronic. Dit is een kunstklep die de aortaklep vervangt. Aanvankelijk gebruikt voor mensen die te ziek waren voor open-hart-operatie, maar geleidelijk aan ook bij mensen die ook dat soort chirurgie konden doorstaan.
De gegevens die Medtronic kreeg van de Society of Thoracic Surgeons/American College of Ccardiology transcatheter valve therapy registry bestond echter uit informatie die niet tot personen herleidbaar was. Het rapport dat Medtronic vervolgens richtin FDA stuurde bevatte daardoor niet die informatie die de FDA had behoren te ontvangen, namelijk op persoonsniveau. In het vierde kwartaal 2018 ging het bijv. om 472 personen.
Identiek
Een vrijwel identiek verhaal gaat op voor de Sapien 3 transcatheterklep van de firma Edwards Lifesciences. De leiding van die firma klaagt er ook over dat de registrerende organisatie onvoldoende informatie levert om op persoonsniveau aan de FDA door te kunnen geven wat er mis was bij de overlijdensgevallen waar hun klep in het geding was.
Onder zelfde protocol vallend
In het artikel staat vermeld dat het gewraakte protocol gaat om zes “devices” in drie registratiedatabases. De schrijfster geeft aan dat het verder gaat om de Mitraclip van de firma Abbott Dat is een klemmetje om een lekkende mitraalklep te repareren. Ook om de Watchman, een “device” om een uitgezet deel van één van de hartboezems op te vullen. Daarnaast om de Medtronic’s Valiant Thoracic Stent Graft, bedoeld om een defect stuk van de aorta te verhelpen en tenslotte om een speciale katheter van Medtronic om plotselinge blokkering van grote slagaderen te verhelpen als die door een groot stolsel acuut verstopt zijn geraakt.
Betekenis
Wat betekent dit voor de beoordeling van de FDA van via de grote vaten ingebrachte kunstkleppen en gelijkende “medical devices”? Het wonderlijke is dat een systeem dat door de FDA bedacht was om snel te kunnen reageren op sterfgevallen en complicaties bij dit soort nieuwe medische ontwikkelingen zich zelf in de staart bijt. Het blijkt dat de registratie eerder moeilijker dan makkelijker is geworden. Daardoor niet transparant is. Het betekent ook dat mogelijk te gevaarlijke nieuwe ontwikkelingen te laat ontdekt worden. En dat was nu juist nooit de bedoeling. Internationaal heeft het ook consequenties omdat vanuit de rest van de wereld het eventueel toelaten, maar ook afwijzen van medische uitvindingen door de FDA nauwgezet in de gaten wordt gehouden. De transparantie van de FDA is dus duidelijk in het geding met het op een dergelijke manier delegeren van verantwoordelijkheden.
Acties FDA
Nadat Kaiser Health News in juni 2019 al meldde dat het mis ging met het delegeren van verantwoordelijkheden bij toelating van medische hulpmiddelen stopte de FDA al met het delegeren van verantwoordelijkheid bij ruim 100 “medical devices”. Daarbij bleven de zes hierboven genoemde buiten schot. Uit het artikel van Jewett blijkt niet dat FDA daar een definitief besluit over genomen heeft. Het is zeer aannemelijk dat het ook voor deze hulpmiddelen wordt afgeschaft.
W.J. Jongejan, 10 januari 2020.
Afbeelding van burlesonmatthew via Pixabay


Recente reacties